La Sindrome di Sturge-Weber
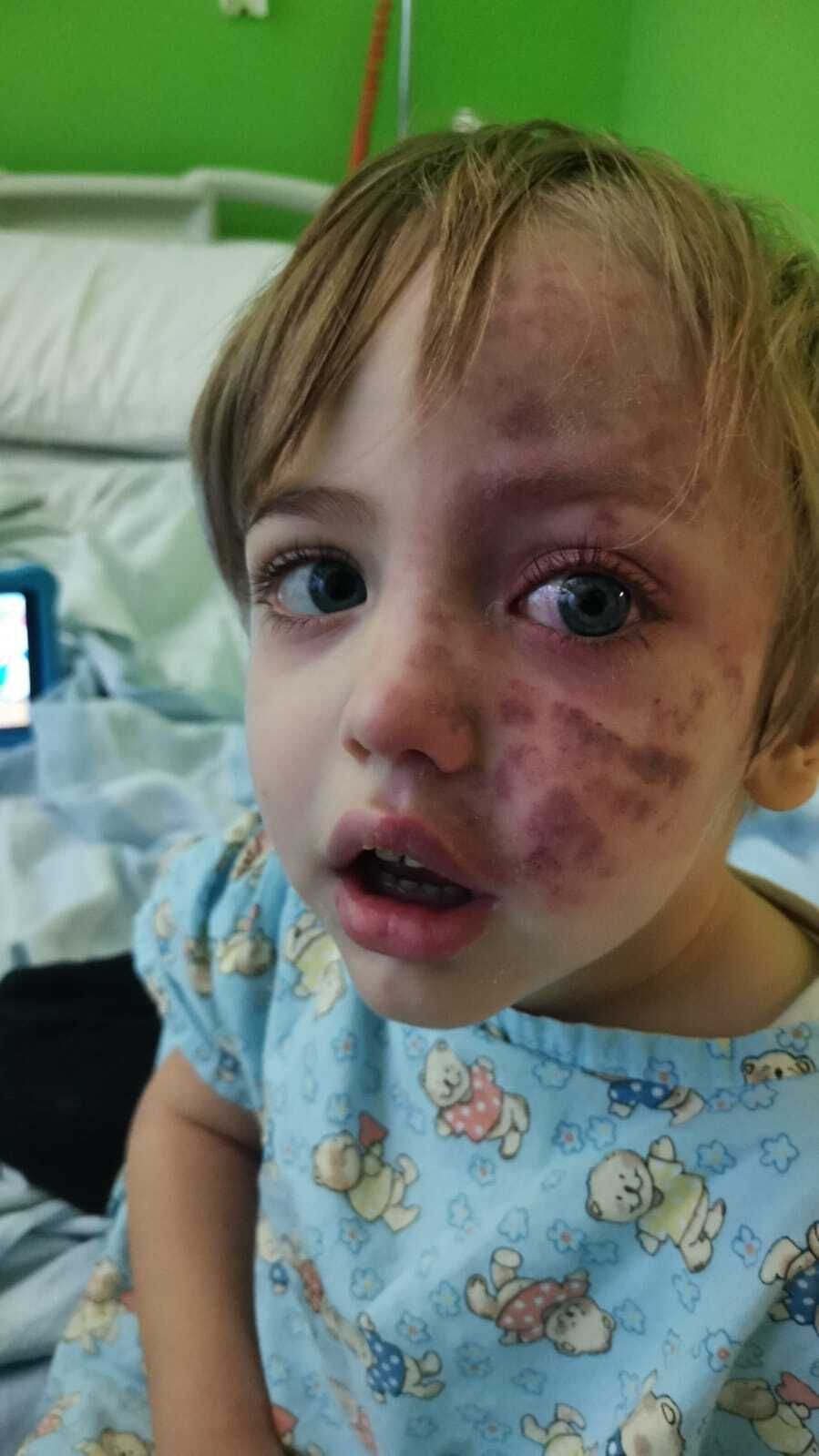
La Sindrome di Sturge-Weber è una malattia neurocutanea. Questo significa che colpisce principalmente la pelle e il sistema nervoso.
La malattia è causata da malformazioni dei vasi capillari e dei piccoli vasi venosi. Queste malformazioni sono anomalie che si formano durante lo sviluppo nell’utero materno.
I vasi capillari sono i vasi sanguigni più piccoli del nostro organismo. I piccoli vasi venosi, invece, raccolgono il sangue dalla periferia dell’organismo e lo portano al cuore.
La prevalenza alla nascita in Europa varia: si stima che sia tra 1/20.000 e 1/50.000. Inoltre, il rischio di SSW nei neonati può variare dal 15% al 40%, a seconda dell’estensione della malformazione facciale.
È essenziale capire la condizione per affrontarla con efficacia.
01
Cenni Storici:
Identificata per la prima volta nel XIX secolo, la ricerca sulla Sindrome di Sturge-Weber ha progredito notevolmente, guidata dalla determinazione scientifica.
02
Manifestazioni:
Oltre alla caratteristica malformazione cutanea, la Sindrome presenta una gamma di sintomi neurologici. Conoscere è il primo passo per intervenire.
03
Evoluzione:
La Sindrome di Sturge-Weber varia tra individui; alcuni sintomi rimangono stabili, altri evolvono nel tempo.
04
Diagnosi:
La diagnosi si basa su specifiche manifestazioni cliniche. Un'attenta osservazione da parte degli specialisti è cruciale per l'identificazione.
05
Terapie e trattamenti:
Diverse opzioni terapeutiche sono disponibili in base alla manifestazione della sindrome. La scelta giusta può fare la differenza.
FAQ (Domande Frequenti)
-
Quali sono le caratteristiche principali della sindrome?
Le caratteristiche principali includono malformazioni vascolari, cerebrali, manifestazioni cutanee come macchie di vino porto e patologie oculistiche come ipertono oculare
-
Qual è la causa della sindrome di Sturge-Weber?
La causa precisa è sconosciuta, ma coinvolge una mutazione genetica spontanea che compare nelle prime settimane di vita intrauterina
-
Quali sono i rischi associati alla sindrome?
I rischi includono crisi epilettiche e ritardi nello sviluppo motorio e cognitivo. Talvolta le crisi sono precedute da episodi di emicrania ed emiplegie transitorie.
-
Come può essere gestita la sindrome nella vita quotidiana?
La gestione coinvolge un approccio multidisciplinare con il coinvolgimento di neurologi, dermatologi e altri specialisti.
-
Quali sono le opzioni di supporto psicologico e sociale disponibili?
Gruppi di supporto, consulenza e risorse online possono offrire supporto emotivo e informazioni pratiche per pazienti e famiglie.
-
Ci sono risorse educative disponibili per pazienti e famiglie?
Organizzazioni mediche e associazioni pazienti forniscono spesso materiale informativo e risorse educative sulla sindrome.
-
Quali sono le ultime ricerche e sviluppi riguardo la Sindrome di Sturge-Weber?
La ricerca è in corso per comprendere meglio la malattia e sviluppare trattamenti più efficaci.
Come Sostenerci
Ogni donazione contribuisce direttamente a sostenere le famiglie, finanziare ricerche e migliorare la vita di chi ha la Sindrome di Sturge-Weber.
Effettua una donazione a:
IBAN: IT58D0883331030000180101323
News


Vuoi rimanere aggiornato sulle nostre iniziative?
Iscriviti alla nostra newsletter e non perderti nessuna novità!
Iscriviti alla nostra newsletter e non perderti nessuna novità!

Associazione Sindrome di Sturge Weber Italia
Altavilla Vicentina (VI) – Via Monico 86,
CAP 36077 |
+ 39 347 0669700
info@sturgeweberitalia.org
|
sturgeweberitalia@pec.it
Codice Fiscale 97807380015 - Iscrizione al registro provinciale ONLUS prot. n° 2016/38702 del 30.05.2016I BAN: IT58D0883331030000180101323
Tutti i diritti riservati Associazione Sindrome di Sturge Weber Italia - Realizzato da PS COMPANY SRL